寄稿論文
含窒素化合物の確実かつ簡便な合成法:
Ns-strategy と高活性トリチル型レジン
静岡県立大学 薬学部 薬品製造化学教室菅 敏 幸
東京大学 大学院薬学系研究科福 山 透
1. はじめに
窒素原子を有する化合物はユニークな生理活性を持つことが多く,効率的合成は医薬品開発 等において重要な課題である。しかし,高極性な含窒素化合物はハンドリングが困難なことが 多く,さらに酸化条件などに不安定なため使用可能な反応が限られている。このため,多くの 官能基が共存する化合物の合成には,窒素原子の保護は必要不可欠であり,その選択は合成の 重要な問題となる。1)我々は,穏和な条件下脱保護可能なニトロベンゼンスルホニル(Ns)基が アルキル化反応の活性化基としても働くことを見いだし,この方法を用いた2級アミン合成法 (Ns-strategy)の開発に成功している。2)本合成法は,発表以来多くの化学者に使われる有用な方 法となり,我々も生理活性天然物の全合成へと展開してきた。3) 本方法論のポリアミントキシ ン合成への展開過程で,我々は市販のトリチルレジンよりも充填効率の高い固相の開発に成功 した。4)このレジンによる固相合成は,水溶性のポリアミンの合成に一切の精製操作を必要とし ない画期的な方法となった。本寄稿論文では,これらの方法論を基盤とする含窒素化合物の簡 便な合成法を紹介する。2. 1 級アミンから 2 級アミンへの合成
アミンを選択的にモノアルキル化することは難しく,そのため1級アミンから対応する2級 アミンへの変換も確実な方法は確立されていない。スキーム1に,代表的な2級アミンの合成 例を示した。1級アミン1 とアルキルハライドやスルホン酸エステルとのアルキル化反応を行 うと,望みの2級アミン2と共に3級アミン3や4級アンモニウム塩4が副生することが問題と なる。また,NaBH3CNを用いたアルデヒドやケトンに対する還元的アミノ化反応も,2級アミ ン5に立体的な制約がない場合は3級アミン6を副生することがある。モノアルキルアミド7に 変換後,LiAlH4や BH3のような強い還元剤を用いる方法は信頼がおけるが,多官能基性化合物 には必ずしも適用できない。最近になり,トルエンスルホン(Ts)アミド8a やトリフルオロア セトアミド8b に対する光延反応5)が開発されているが,脱保護(9a,b → 2)に強い塩基性条件 を必要とするため,本条件に不安定な官能基を有する2級アミン合成には適当ではない。 * 便宜上,ニトロベンゼンスルホンアミド誘導体の 2- ニトロ体を Ns,4- ニトロ体を pNs,2,4- ジニトロ体 を DNs と略させていただく。Scheme 1. Conversion of primary amines to the corresponding secondary amines.
2 . 1 . 2- あるいは 4- ニトロベンゼンスルホンアミド
2) スルホンアミドは強酸性条件下や通常の塩基性条件下で安定なため,信頼のおける窒素原子 の保護基である。さらに,カルバメートやアミドにしばしば見られる1H NMRシグナルのブロー ド化がないことも利点の一つである。しかし,汎用されてきた Ts や Ms 基は脱保護に過酷な条 件が必要である。これに対し,我々は,ニトロベンゼンスルホンアミド*が極めて穏和な条件で アミンへと変換可能であることを見いだした。スキーム 2 に,Ns 基を保護・活性化基として用 いた1級アミンから2級アミンへの合成法(Ns-strategy)の代表例として,p- メトキシベンジル アミン(10)の反応を示した。 R2 X base R1 NH2 R1 N R1 N R1 N R2 R2 R2 H R2 R2 R2 X 1 2 3 4 R2 CHO reductive amination R1 NH2 R1 N R1 N H 1 5 6 R2 R2 R2 R2 CO2H condensation R1 NH2 R1 N H 1 7 R2 O reduction R1 N H 5 R2 R2 OH Mitsunobu conditions R1 H N R1 N R2 P 9a: P = Ts 9b: P = CF3CO deprotection R1 N R2 H 2 P 8a: P = Ts 8b: P = CF3COScheme 2. Conversion of primary amines to the corresponding secondary amines via Ns-strategy.
OMe
NH2
OMe
NH R
primary amine: 10 secondary amine: 15
SO2Cl X Y SR' X Y a; X = NO2, Y = H b; X = H, Y = NO2 c; X = NO2, Y = NO2 (Ns) (pNs) (DNs) 11 Base SO2, OMe HN SO2 X Y OMe N SO2 X Y OMe N SO2 X Y R R R'S 12 13 14 RX or ROH R'S
1級アミン10 を,塩基存在下 2- ニトロベンゼンスルホニルクロリド(NsCl)11a と反応させ るとスルホンアミド12a が得られる。この N- モノ置換スルホンアミド 12a は,ハライド(R-X) とのアルキル化反応,あるいはアルコール(R-OH)との光延反応6)の条件にてアルキル化が容 易に進行し,N- ジ置換スルホンアミド13a を与える。ニトロベンゼンスルホンアミドは,その 小さな pKaからアルキル化反応の活性化基として作用し,Ts アミドでは比較的困難な光延反応 も円滑に進行する。13a に対して,塩基存在下 PhSH の様なソフトな求核剤(Nu−)を作用させ ると,芳香環にチオラートアニオンが求核付加してマイゼンハイマー複合体14aを形成後,SO2 の脱離を伴い2級アミン15 へと変換される。このように Ns 基は穏和な条件にて脱保護が可能 であるが,強い酸性条件や塩基性条件にも安定であるため,種々の合成反応に耐えうる1級あ るいは2級アミンの保護基でもある。さらに,これら一連の反応過程では一切のラセミ化が進 行しないことも確認している。また,4- ニトロベンゼンスルホニルクロリド(pNsCl:11b)も 11a と同様の反応性を有しているが,我々の研究室では,安価な NsCl 11a を多用している。** (東京化成工業 NsCl: N0142,pNsCl: N0144)
2 . 2 . 2, 4- ジニトロベンゼンスルホンアミド(DNs)
7) 2,4- ジニトロベンゼンスルホニル(DNs)基も,Ns 基と同等のアルキル化能を有しているが, 塩基存在下での長時間加熱に不安定であるという欠点を有している。しかし,Ns 基より穏和な 条件で脱保護可能なため,DNs 基のみを選択的に除去することが可能である。スキーム 3 に 示した様に,2つのニトロベンゼンスルホニル基で保護されたジアミン16のDNs基選択的な除 去は(HSCH2CO2H, Et3N)の条件にて可能で,2級アミン17 が定量的に得られた。また,この方 法は副生する2,4-ジニトロフェニルチオ酢酸がEt2Oと飽和重曹水との分液操作で除ける利点も ある。Scheme 3. Selective deprotection of DNs group.
3. 保護1級アミンの合成
(N- カルボアルコキシニトロベンゼンスルホンアミド)
8) アルキルハライドやアルコールに対し求核置換反応により窒素原子を導入し,1級アミンを 合成する方法も有用な反応である。これまで,ガブリエル法9)やアジドを用いる方法が開発され ているが,保護された1級アミンを直接合成する優れた方法はあまりない。最近になり, Weinrebらの TsNHBoc に対する光延反応5)や,角田らの TsNH2に対する改良光延反応10)が報告 されている。我々は,Ns 基の特徴を利用することで,さらに有用な窒素求核剤を開発できると 考えた。アンモニア(NH3)と NsCl(11a)から容易に調製可能な NsNH2(18)は,Et3Nと触媒量の DMAP 存在下カルバメート化が進行し,Boc 体19a が結晶として得られる。このスルホ ンアミド19a は,アルキルハライドとのアルキル化反応やアルコールとの光延反応が円滑に進 行する。代表例として,スキーム 4 に 19a と(−)- 乳酸エチル(20)との反応を示した。通常の 光延反応の条件下にて得られる21 の,Ns 基と Boc 基は区別して脱保護が可能である。すなわ ち,21 にチオールを作用させると,Ns 基の脱保護が進行し N-Boc アラニン誘導体 22 が得られ る。また,21 の Boc 基の除去は酸性条件にて進行し,Ns アミド 23 への変換も可能である。 Bn N N PMB DNs Ns Bn N N PMB H Ns HSCH2CO2H Et3N CH2Cl2 (99%) 16 17 ** 2- ニトロベンゼンスルホンアミドでは問題がないが,4 - ニトロ体の脱保護には副反応を伴うことが 報告されている:P. G. M. Wuts, J. M. Northuis, Tetrahedron Lett., 39, 3889 (1998).
22 の Boc 基を除去すると1級アミンが得られ,23 は Ns-strategy により2級アミンへの変換が可 能である。さらに,スルホンアミドの Alloc 体19b や Cbz 体 19c も同様に合成が可能であり,こ れら結晶性のアンモニア等価体であるスルホンアミド18と19a-cは東京化成工業より市販され ている。
Scheme 4. Alkylation and deprotection of N-Boc-Ns-amides.
4. 分子内アルキル化反応(中員環化合物の合成)
11) 最近,オレフィンメタセシス反応をはじめとする大中員環化合物の合成研究が報告されてい るが,含窒素複素環合成に直接的な窒素求核剤を用いるものは少ない。我々は,Ns 基のアルキ ル化を分子内反応に展開したところ,通常困難である8,9,10員環の環化にも有効である ことを見いだした。スルホンアミド18 から合成した,ハライド 24 に n-Bu4NI存在下 Cs2CO3を 作用させると,環化反応は円滑に進行し25 が得られた。同様に,アルコール 26 も光延反応の 条件に付すことで,環化体25 が得られた。環化前駆体の 24 や 26 は反応を有利にする置換基を 有していない。さらに本反応は,高希釈条件を必要としないことも特徴である。 NsNH2 H N Ns R EtO2C OH Me Boc2O, Et3N DMAP (0.1 eq) CH2Cl2 (95%) DEAD, PPh3 benzene (91%) 21 18 19a: R = Boc 19b: R = Alloc 19c: R = Cbz 20 EtO2C N Boc Me Ns EtO2C NH2 Me TFA 22 EtO2C N H Boc Me HSCH2CO2 K2CO3 DMF TFA (100%) Ns-strategy EtO2C NH Me R 23 EtO2C N H Ns Me 21 (94%)Scheme 5. Construction of medium-sized rings.
5. 天然物ポリアミントキシンの合成
近年の微量分析技術の進歩に伴い,ポリアミン鎖を有する天然物が数多く単離されている。12) これらの多くは,強力な生理活性を有しながらも天然からは極微量にしか得られないことが多 い。そのため,多くの合成研究が行われているが,2級アミンの構築に関しては満足のいくも のはあまりない。我々は,Ns-strategy を用いることで効率的な合成が可能であると考え,天然物 ポリアミン類の合成に着手した。 NsHN Br n NsN n NsHN HO n Cs2CO3 n-Bu4NI DEAD PPh3 n = 1: 62% n = 2: 64% n = 3: 66% n = 1: 59% n = 2: 57% n = 3: 62% 24 25 265 . 1 . ジアミンの選択的保護
ジアミンの二つのアミノ基を区別した化合物は,ポリアミン合成の有用な素子となるだけで なく,プローブ分子等のリンカーとしても活用できる有用な化合物である。しかし,一般に対 称ジアミンのモノカルバメート化は収率が低いうえに,精製も煩雑である。13)我々は,ジアミン のNs化が一方の窒素のみに選択的に進行することを見出した。 スキーム6に示したように,1,3-ジアミノプロパンに,低温下,NsCl をゆっくり加えていくと,モノスルホニル化反応が選択的 に進行した。モノスルホニル化されたジアミン27の単離は,その塩酸塩をNaOEtで中和後,NaCl をろ過,溶媒を留去,過剰の未反応のジアミンを減圧留去することで可能である。さらに,n = 2,3 いずれのジアミンからも,モノスルホニル体28,29 が高収率で得られる。これらのジアミ ン誘導体(n = 1-5)も東京化成工業から市販されている。Scheme 6. Selective mono-nosylation of diamines.
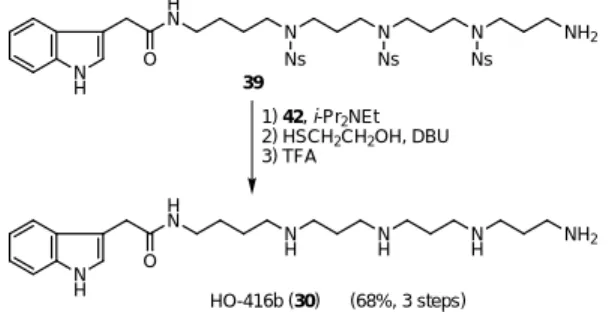
5 . 2 . HO-416b(30)の全合成
5)クモの毒腺から単離されたポリアミンは,興奮性グルタミン酸受容体を特異的に阻害するこ とが知られている。14)さらに,その受容体は脳神経細胞の記憶や学習の働きと密接に関わるた め,医薬や農薬の開発のためのリード化合物として期待されている。15)我々は,Ns-strategy によ りポリアミンの簡便な合成が可能となると考え,クサグモ Holoena curta から単離された
HO-416b (30)16)の合成研究に着手した。 H2N n NH2 H2N n NH Ns NsCl (11a), EtOH -20 °C; NaOEt 27: n = 1 (83% from NsCl) 28: n = 2 (77% from NsCl) 29: n = 3 (87% from NsCl) N H H N N H NH O N H NH2 HO-416b (30)
Fig. 1. Structure of HO-416b (30).
合成はスキーム 7 に示した様に,左右のフラグメントに分け最後に連結する収束的な方法を 用いた。左のフラグメント32 は,インドール酢酸 31 とジアミン 28 との縮合により合成した。 右のフラグメントは,27 を Boc 化したジアミン 33 を原料とした。33 と過剰量の 1,3- ジブロモ プロパンを作用させ臭素体34 とし,さらにスルホンアミド 35 とのアルキル化反応により 36 を 合成した。32とのカップリングはアルコール36との光延反応でも可能であるが,精製が容易な ハライドによるアルキル化を用いることとした。一度アルコール36 を Ms 化後,ヨウ素体 37 へ と変換し,32 とのカップリングを試みた。反応は塩基性条件下,円滑に進行しカップリング体 38 を得ることができた。さらに Boc 基の脱保護を行い1級アミン 39 へと導いた。
Scheme 7. Total synthesis of HO-416b (30). ここで,39 の Ns 基の脱保護により,HO-416b(30)が得られるが,Ns 基の脱保護には問題 がないものの,30 の単離精製に困難が伴った。通常,水溶性のポリアミン類の精製には,逆相 HPLC やイオン交換樹脂が用いられる。しかし,固相上で Ns 基の脱保護を行えば,洗浄による 過剰の試薬や副生するニトロベンゼン誘導体の除去が可能となり,精製操作が必要ないと考え た。まず,1級アミン39 の担持を比較的高価な 2- クロロトリチルレジン 40 にて検討したが充 填効率は満足のいくものではなかった。そこで,充填効率向上のため,反応点がポリマーから 離れたレジン42 を設計した(スキーム 8)。42 は安価な Merrifield resin を p- ヒドロキシトリチ ルアルコールでアルキル化後,トリチルアルコール41をクロリドに変換することで調製可能で ある。この42 は,反応点のトリチルカチオンをパラ位の酸素原子が安定化していることからも 高い反応性を有している。さらに,このレジンは切り出し後,クロロ化の条件に付すことで何 度も再利用可能である。このレジン42 の安定前駆体である 41 も東京化成工業から市販されて いる。 N H OH O N H H N O NH Ns PivCl, Et3N; 28, Et3N, DMAP (97%) 31 32 HN NH R Ns Br Br K2CO3 (97%) N NH Boc Ns Br 34 27: R = H 33: R = Boc (99%) Boc2O, Et3N HO NH Ns N NH Boc Ns N 36: X = OH 37: X = I 1) MsCl, Et2) NaI, 2-butanone3N Ns X 34, Cs2CO3 n-Bu4NI (86%) 35 32 + 37 N NH R Ns N Ns N Ns H N N H O Cs2CO3 (94%) 38: R = Boc 39: R = H SOCl2, MeOH (96%) (98%)
Scheme 8. Synthesis of trityl-type resin (42). Cl P X O Cl P Cl P OH HO 40 41: R = OH 42: R = Cl SOCl2 K2CO3 Merrifield resin
スキーム 9 に示した様に,使用直前に調製した固相 42 に i-Pr2NEtを用い39 を担持した。固相
上でのNs基の脱保護は(HSCH2CH2OH, DBU)の条件にて行った。過剰の試薬等を洗い流しレジ ンを乾燥後,(1% TFA-CH2Cl2)にて切り出した。その溶媒を留去することで純粋なHO-416b(30) の TFA 塩が得られた。このように,最後の脱保護を固相上で行うことで,高極性なポリアミン の単離に一切の精製操作が必要なかった。
Scheme 9. Completion of total synthesis of HO-416b (30).
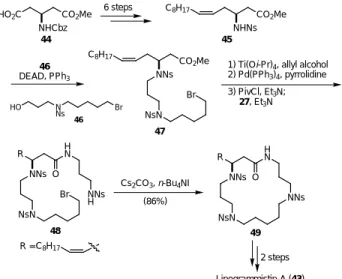
5 . 3 . 18 員環ポリアミン lipogrammistin-A(43)の全合成
17) 大環状ポリアミンを有する天然物も植物由来のアルカロイドなど数多く知られている。18)これ らも多くの合成研究があるが,いずれも大員環構築に満足のいくものではない。我々は,中員 環合成に有効であったNs基の分子内アルキル化反応が,大環状アミン合成にも有効であると考 え Lipogrammistin-A(43)の合成に着手した。 N NH2 Ns N Ns N Ns H N N H O N H NH2 N H N H H N N H O 39 1) 42, i-Pr2NEt 2) HSCH2CH2OH, DBU 3) TFA HO-416b (30) (68%, 3 steps)Fig. 2. Structure of Lipogrammistin-A (43).
Lipogrammistin-A(43)は,橘,伏谷らの共同研究によって,ヌノサラシ科の魚の皮膚から 単離構造決定されたポリアミントキシンである。19)この化合物は18員環のマクロラクタム環と 長い脂肪鎖を有するβ-アミノ酸から成る特異な構造を有している。我々のNs基の分子内アルキ ル化反応を鍵工程とする全合成の概略をスキーム 10 に示した。β- アミノ酸誘導体45 は,文献 既知の光学活性なカルボン酸4420)より Wittig 反応を鍵工程として用い 6 段階にて合成した。ス ルホンアミド45 とアルコール 46 とのカップリングは,光延反応により行い 47 が得られた。47 のメチルエステルに塩基性加水分解を行なうとアルキルスルホンアミドのβ脱離を伴うため,ア リルエステルに変換後,Pd触媒により脱保護を行った。カルボン酸とジアミン誘導体27を混合 酸無水物法により縮合し環化前駆体48とした。鍵となる18員環構築は,(Cs2CO3, n-Bu4NI) の 条件により達成した。最後に49 の3つの Ns 基を全て脱保護し,2- メチル酪酸を反応させるこ とで Lipogrammistin-A(43)の全合成を達成した。 NH O H N N N O O Lipogrammistin-A (43)
Scheme 10. Total synthesis of Lipogrammistin-A (43).
5 . 4 . Ephedradine A (50) の全合成
21)Lipogrammistin-A(43)全合成の好結果により,さらに構造が複雑かつ合成に困難であると予 想された Ephedradine A(50)の合成研究に着手した。Ephedradine A(50)は中国伝承薬の麻黄 根の活性成分として単離された,スペルミンアルカロイドである。この化合物は,1979 年に東 北大学の曵野らにより構造決定された古くから知られる化合物である。22)しかし,その合成例は 1985 年の Wasserman らによる O- メチル体である51 のラセミ体の報告のみである。23)50 の合成 の困難な点は,酸に不安定なジヒドロベンゾフラン環や塩基に不安定なβアミノ酸誘導体の存 在下の,2つのマクロラクタム環の構築が挙げられる。この点,Ns-strategyは穏和な条件にて環 化と脱保護が進行するため,Fig. 3に示したように,コア部分52 を光学活性体として合成した 後,ポリアミン骨格を構築することが可能である。 Lipogrammistin-A (43) HO2C CO2Me NHCbz CO2Me NHNs C8H17 6 steps 44 45 46 DEAD, PPh3
1) Ti(Oi-Pr)4, allyl alcohol 2) Pd(PPh3)4, pyrrolidine 3) PivCl, Et3N; 27, Et3N CO2Me NNs C8H17 NsN Br 47 HO N Ns Br 46 NNs C8H17 NsN Br H N O NNs H R R = 48 Cs2CO3, n-Bu4NI (86%) NNs NsN H N O NNs R 49 2 steps
Fig. 3. Structure of (-)-Ephedradine A (50).
スキーム 11 に示した,重要中間体 52 の光学活性なジヒドロベンゾフラン環は不斉 C-H 挿入 反応により合成した。24)また,β-アミノエステルはSharplessの不斉アミノヒドロキシル化反応25) を鍵工程として構築した。スルホンアミド52 に対しアルコール 53 を導入した後,窒素原子上 の保護基を Cbz 基に変換し54 とした。続いて TBS 基を除去して得られるアルコールに対して O OR H H N O N H HN H N H O O OBn H H N O OAc CO2Me NsHN OTBDPS 52 (−)-Ephedradine A (Orantine): R = H (50) O-Methylorantine: R = Me (51)
NsNH2(18)を導入後,TBDPS 基を除去して環化前駆体 55 を得た。55 に DEAD と PPh3を作用 させたところ,鍵となる16員環閉環反応が円滑に進行して環化体56が良好な収率で得られた。 続いて,56 のアセトキシ基をアジドへと変換後,メチルエステルをペンタフルオロフェニル (PfpOH)エステル57 とした。続いて,57 に対して PPh3を作用させると Staudinger 反応 26)が進 行し,生じたイミノホスホラン58 と活性エステルによる分子内 aza-Wittig 反応によってイミノ エーテル59 が得られた。これに対して加水分解を行い,マクロラクタム 60 へと変換すること ができた。最後に60 の Ns 基を脱保護後,ベンジル基と Cbz 基を BCl3にて同時に脱保護し, Ephedradine A(50)の全合成を達成した。本全合成は,Ns-strategy の強力かつ多官能基共存性 を明らかにすると共に,アミド結合の形成に新しい化学を見いだすことができた。27)
Scheme 11. Total synthesis of (-)-Ephedradine A (50).
6. 高活性なトリチル型レジン(42)による固相合成
我々が開発したトリチル型固相42は,アミンとの高い充填効率だけでなく,連結した状態(固 相上)での Ns アミドとアルキルハライドとのアルキル化や,アミド化反応,さらには2級アミ ンとの反応も可能なため,以下のコンビナトリアル合成への展開も可能となった。 O H H R O NsHN H Ar CO2Me O H H R O CbzN H Ar CO2Me OTBS N OAc OTBDPS R = 1) DEAD, PPh3 2) KOH, PhSH 3) Cbz-Cl, Et3N (80%) HO OTBS 53 52 54 CbzN H CO2Me NHNs O H H N O OAc HO Ar OBn Ar = DEAD, PPh3 1) CSA, MeOH 2) 18 3) HF (79%) PPh3, DEAD (77%) CbzN H CO2Me O H H N O OAc Ar N Ns 56 55 CbzN H CO2Pfp O H H N O N3 Ar N Ns 57 1) K2CO3 2) MsCl, Et3N 3) NaN3 4) LiOH 5) PfpOH (71%) PfpOH = pentafluorophenol H N O N Ns N PfpO O PPh3 CbzN 58 PPh3 H N O N CbzN 59 N Ns PfpO H N O N CbzN 60 N H Ns O H Ar H O H2O (73%) 1) KOH PhSH 2) BCl3 (73%) 50 WSCD6 . 1 . PhTX-343 (61) の固相合成
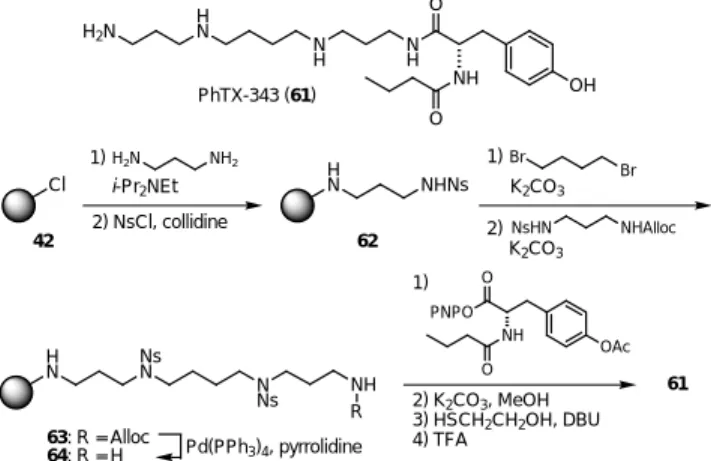
28) スペルミンやスペルミジンに代表されるポリアミン類は,生体内に多く存在する生理活性物 質であり,これらを含む化合物も数多く知られている。29)そのため,多様な化合物を簡便に合成 する方法が強く望まれている。固相42 は最終段階に精製の段階を必要としない,さらに固相上 でのアルキル化が可能となればコンビナトリアル合成も容易となる。そこで,スペルミン誘導 体の Philanthotoxin-343(PhTX-343:61)30) の合成に着手した。合成の概略を,スキーム 12 に 示した。ジアミノプロパンをレジン42 に連結させた後,遊離のアミンを Ns 基で保護し Ns アミ ド62 とした。さらに,ジブロモブタン,スルホンアミドを順次アルキル化することで,保護さ れたスペルミン誘導体63 を簡便に合成できた。この方法は,様々なジアミンやハライドを組み 合わせることも可能であり,さらにアルコールとの光延反応も容易であるため,ポリアミン鎖 の長さを変えたライブラリーも容易に合成可能である。また,Alloc 基を除去した64 は,その 末端アミンへのアルキル化やアシル化が容易であるため,スペルミン連結体の合成にも有用な 中間体である。PhTX-343(61)の合成は,64 にチロシン誘導体を導入した後,Ns 基を脱保護 し固相から切り出すことで,レジン42 から 9 段階,総収率 75%にて全合成を達成した。Fig. 4 には,最終段階の溶媒を留去しただけの1H NMRを示した。本合成は,すべての段階に一切の 精製を行なっていないにも関わらず,Fig. 4に示した純品が得られる。このように,我々の固相 樹脂42 と Ns-strategy を組み合わせることで画期的な合成法の開発に成功した。Scheme 12. Solid-phase synthesis of PhTX-343 (61).
Fig. 4. 1H NMR spectra of synthetic PhTX-343 (61). H2N H N N H NH O NH O OH PhTX-343 (61) Cl HN NHNs H N NsN N Ns NH R H2N NH2 Br Br NsHN NHAlloc K2CO3 K2CO3 1) 2) i-Pr2NEt 2) NsCl, collidine 1) PNPO O NH O OAc 1) 2) K2CO3, MeOH 3) HSCH2CH2OH, DBU 4) TFA 61 63: R = Alloc 64: R = H Pd(PPh3)4, pyrrolidine 42 62
6 . 2 . ジペプチド型γ - セクレターゼ阻害剤 DAPT 誘導体のパラレル合成
31) 我々の研究室では,数年前より本研究科の共同研究プロジェクトとして,アルツハイマー病 の発症に重要なγ- セクレターゼ阻害剤32)の開発とその機能解明の研究を展開している。33)その 一環として,Elan 社のグループが阻害剤として報告しているDAPT(65)34)の構造活性相関研究 を行い,C 末端 t-Bu エステルをアミドに変換した化合物は65 と同程度の活性を保持しているこ とを見いだした。Fig. 5. Structure of DAPT (65).
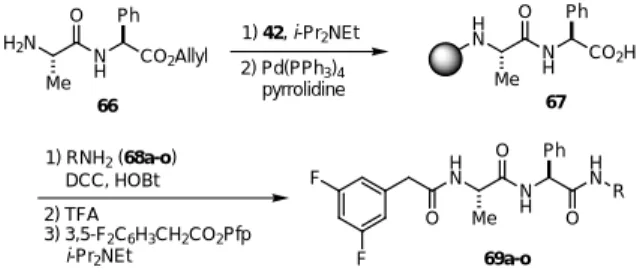
そこで,DAPT(65)の C 末端の多様なアミド化合物を簡便に合成するため,我々が開発した 固相樹脂42 を用いたパラレル固相合成を試みた。ジペプチド 66 を固相樹脂 42 へ担持した後, アリルエステルを除去してカルボン酸67 とした。67 に対する各種アミン(68a-o)との縮合は 過剰の HOBt 存在下 DCC を用いることにより,円滑に進行した。続いて酸性条件下固相樹脂よ り切り出し,活性エステル法にてフェニル酢酸セグメントを導入した。反応終了後,粗生成物 の結晶をろ取・洗浄するのみで目的とする DAPT(65)の C 末端をアミドに変換した誘導体 (69a-o)を得ることに成功した。
R = n-C7H15 (a), CH2Ph (b), CHPh2 (c), CH2C6H4-o-OMe (d), CH2C6H4-m-OMe (e), CH2C6H4-p-OMe (f), CH2C6H4-o-Cl (g), CH2C6H4-m-Cl (h), CH2C6H4-p-Cl (i), CH2C6H4 -o-Br (j), CH2C6H4-m-Br (k), CH2C6H4-p-Br (l), CH2C6H4-p-F (m), CH2C6H4-p-CO2Me (n), CH2C6H4-p-Bz (o).
Scheme 13. Parallel synthesis of DAPT derivatives (69a-o).
通常,Merrifield 法に代表されるペプチドの固相合成は,C 末端を固相に連結し N 保護アミノ 酸との縮合と脱保護を繰り返し行われる。それに対し,本合成法は N 末端を連結させた N- アシ ルアミノ酸にも関わらず,α位のエピマー化を伴うことなく縮合が進行する。そのため,N 末端 から C 末端へのペプチド鎖の伸長も可能である。また,DAPT のアミド誘導体(69a-o)の中で, 69o は DAPT(65)の約 30 倍強力な活性を有していたため,プローブへの展開に有効となった。 F F H N O Me N H O Ph CO2t-Bu
N-[N-(3,5-difluorophenylacetyl)-L-alanyl]-(S)-phenylglycine-tert-butyl ester (DAPT 65)
F F H N O Me N H O Ph H2N Me N H O Ph CO2Allyl 66 1) 42, i-Pr2NEt 2) Pd(PPh3)4 pyrrolidine H N Me N H O Ph CO2H 67 1) RNH2 (68a-o) DCC, HOBt 2) TFA 3) 3,5-F2C6H3CH2CO2Pfp i-Pr2NEt H N O R 69a-o
6.3.フォトアフィニティプローブの固相合成
35) 近年の家族性アルツハイマー病の研究により,疾患原因遺伝子の一つとしてプレセニリン (PS)がクローニングされた。その後,PS 遺伝子が産出する膜タンパクを中心とする,巨大複 合体がγ- セクレターゼの本体であることも明らかになった。36) しかし,膜プロテアーゼの生化 学的解析は困難であるため,フォトアフィニティラベリング法が数少ない有効な解析手段であ る。37) プローブ合成には,タンパクと高い親和性(強い活性)を有する化合物に光照射により 架橋する光反応性基の導入が必要となる。ベンゾフェノンはフォトアフィニティラベリングの 際の光反応性基として多用される。そのため,69o の活性が DAPT(65)より増強されることは 好都合であった。さらに,ビオチンタグの導入はアビジンとの高い親和性を利用した,簡便な タンパクの検出や精製が可能となる。そこで我々は,光反応性基のベンゾフェノンとビオチン を,同時にかつ簡便に導入可能な固相樹脂70 を合成した。 前述の方法で合成したモノNsジアミンをリンカーとして用い,光延反応によりベンゾフェノ ンと連結,ビオチンを縮合後,Ns 基の脱保護により2級アミンを合成した。得られた2級アミ ンを固相樹脂42に担持し末端に,リガンドと反応するアミノ基を有するベンゾフェノン−ビオ チン一体型固相樹脂70 の合成に成功した。続いて,DAPT(65)のカルボン酸誘導体とアミン 70との縮合後,酸性条件下固相から切り出すことにより,フォトアフィニティプローブ71の合 成に成功した。この場合も,30 や 61 の合成の場合と同様に最終段階に煩雑な精製を必要とし なかった。また,ビオチンが導入された化合物は,有機溶媒に不溶となるため精製に苦労する ことが多い。そのことからも,この合成戦略の意義は大きいと考えている。現在,プローブ71 を用いたフォトアフィニティラベリングの実験により,DAPT(65)存在下にて消失する,プレ セニリンに由来する 23 および 26 kDa のタンパクの検出に成功している。38)Scheme 14. Solid-phase synthesis of photoaffinity probe (71).
7. おわりに
医薬品等の生体で機能を発揮する化合物の多くは,媒体が水のためその溶解度は重要かつ深 刻な問題となる。一般に,窒素原子を有する化合物は水溶性が高いことから多くの医薬品に存 在する。しかし,本論文の冒頭で述べたように含窒素化合物の合成には困難が伴うことが多い ため,敬遠する合成化学者も多いと聞く。私達は,Ns 基がそれらの問題を解決してくれると信 じ(願い)本研究を広範に展開してきた。その過程で,Ns 基は保護(守備)とアルキル化の活 性化(攻撃)の両方に活躍をすることを見いだし,いくつかの天然物の全合成とアルツハイマー 病など医薬の開発への貢献が可能となった。最後に,本寄稿論文にて紹介した化学を多くの方 に使っていただけると著者らの最も大きな喜びとなる。 F F H N O Me N H O Ph H N O H N HN O NH HN S O H H 8 3 Biotin-tag Cross-linking group LinkerH2N O N HN O NH HN S O H H n 3 O 70: n = 8 71
以上,本寄稿論文で紹介してきた研究は東京大学大学院薬学系研究科天然物合成化学教室に おいて遂行されたものであり,文献に記載したすべての学生の献身な努力に深謝する。また,後 半に紹介したγ- セクレターゼに関する研究は,本研究科の臨床薬学教室と創薬理論科学教室と の共同研究であり,筆者らに天然物の全合成以外の分野の研究に挑戦する機会を与えてくれた 岩坪威教授と夏苅英昭教授に感謝する。
参考文献
1) T. W. Green, P. G. Wuts, “Protective Groups in Organic Synthesis” 3rd ed., John Wiley and Sons, Inc., New York, 1998, p. 494.
2) (a) T. Fukuyama, C.-K. Jow, M. Cheung, Tetrahedron Lett., 36, 6373 (1995). (b) W. Kurosawa, T. Kan, T. Fukuyama, Org. Synth., 79, 186 (2002).
3) Ns-strategyの総説 : (a) 菅 敏幸 , 福山 透 , 有機合成協会誌 , 59, 779 (2001). (b) T. Kan, T. Fukuyama,
Chem. Commun., 2004, 353.
4) (a) Y. Hidai, T. Kan, T. Fukuyama, Tetrahedron Lett., 40, 4711 (1999). (b) Y. Hidai, T. Kan, T. Fukuyama,
Chem. Pharm. Bull., 48, 1570 (2000).
5) (a) J. R. Henry, L. R. Marcint, M. C. McIntosh, P. M. Scola, G. D. Harris, S. M. Weinreb, Tetrahedron
Lett., 30, 5709 (1989). (b) T. Tsunoda, J. Otsuka, Y. Yamashita, S. Ito, Chem. Lett., 1994, 539.
6) 光延反応の総説 : (a) O. Mitsunobu, Synthesis, 1981, 1. (b) D. L. Hughes, Org. React., 42, 335 (1992).
7) T. Fukuyama, M. Cheung, C.-K. Jow, Y. Hidai, T. Kan, Tetrahedron Lett., 38, 5831 (1997). 8) T. Fukuyama, M. Cheung, T. Kan, Synlett, 1999, 1301.
9) Gabriel法の総説 : M. S. Gibson, R. W. Bradshaw, Angew. Chem., Int. Ed., 7, 919 (1968).
10) (a) T. Tsunoda, H. Yamamoto, K. Goda, S. Ito, Tetrahedron Lett., 37, 2457 (1996). (b) 角田らの方法の最 新の総説 : TCI メール , 123, 2 (2004).
11) (a) T. Kan, H. Kobayashi, T. Fukuyama, Synlett, 2002, 697. (b) T. Kan, A. Fujiwara, H. Kobayashi, T. Fukuyama, Tetrahedron, 58, 6267 (2002).
12) 天然物のポリアミンの総説: A. Guggisberg, M. Hesse, “The Alkaloids”, Vol. 50, eds. by Cordell G. A.,
Brossi H. S., Academic Press, New York, 1998, p. 219. 13) W. J. Fiedler, M. Hesse, Helv. Chim. Acta, 76, 1511 (1993).
14) クモ毒の総説(単離,構造決定,合成): A. Schafer, H. Benz, W. Fiedler, A. Guggisberg, S. Bienz,
M. Hesse, “The Alkaloids”, Vol. 45, ed. by Cordell G. A., Academic Press, New York, 1994, p. 1.
15) クモ毒の総説(薬理): A. L. Mueller, R. Roeloffs, H. Jackson, “The Alkaloids”, Vol. 46, eds. by Cordell
G. A., Brossi H. S., Academic Press, New York, 1994, p. 63.
16) G. B. Quistad, C. C. Reuter, W. S. Skinner, P. A. Dennis, S. Suwanrumpha, E. W. Fu, Toxicon., 29, 329 (1991).
17) A. Fujiwara, T. Kan, T. Fukuyama, Synlett, 2000, 1667.
18) 環状ポリアミンの総説 : A. Guggiserberg, M. Hesse, “The Alkaloids”, Vol. 22, 1983, p. 85.
19) (a) H. Onuki, K. Tachibana, N. Fusetani, Tetrahedron Lett., 34, 5609 (1993). (b) H. Onuki, K. Ito, Y. Kobayashi, N. Matsumori, K. Tachibana, N. Fusetani, J. Org. Chem., 63, 3925 (1998).
20) M. Ohno, S. Kobayashi, T. Iimori, Y. -F. Wang, T. Izawa, J. Am. Chem. Soc., 103, 2405 (1981). 21) W. Kurosawa, T. Kan, T. Fukuyama, J. Am. Chem. Soc., 125, 8112 (2003).
22) (a) M. Tamada, K. Endo, H. Hikino, C. Kabuto, Tetrahedron Lett., 20, 873 (1979) (b) P. Dätwyler, H. Bosshardt, S. Johne, M. Hesse, Helv. Chim. Acta, 62, 2712 (1979).
23) H. H. Wasserman, R. K. Brunner, J. D. Buynak, C. G. Carter, T. Oku, R. P. Robinson, J. Am. Chem. Soc.,
107, 519 (1985).
24) W. Kurosawa, T. Kan, T. Fukuyama, Synlett, 2003, 1028.
25) G. Li, H. H. Angert, K. B. Sharpless, Angew. Chem. Int. Ed., 35, 2813 (1996). 26) H. Staudinger, J. Meyer, Helv. Chim. Acta, 2, 635 (1919).
27) H. Fuwa, Y. Okamura, Y. Morohashi, T. Tomita, T. Iwatsubo, T. Kan, T. Fukuyama, H. Natsugari, Tetrahedron
28) T. Kan, H. Kobayashi, T. Fukuyama, Synlett, 2002, 1338.
29) ポリアミンとその連結体の総説 : (a) G. Karigiannis, D. Papaioannou, Eur. J. Org. Chem., 65, 1841
(2000). (b) V. Kuksa, R. Buchan, Synthesis, 2000, 1189.
30) A. T. Eldefrawi, M. E. Eldefrawi, K. Konno, N. A. Mansour, K. Nakanishi, E. Oltz, P. N. Usherwood,
Proc. Natl. Acad. Sci. USA, 85, 4910 (1988).
31) T. Kan, Y. Tominari, K. Rikimaru, Y. Morohashi, H. Natsugari, T. Tomita, T. Iwatsubo and T. Fukuyama,
Bioorg. Med. Chem. Lett., 14, 1983 (2004).
32) セクレターゼに関する最近の総説 : M. S. Wolfe, Nature. Rev. Drug. Discov., 1, 859 (2002).
33) Y. Takahashi, I. Hayashi, Y. Tominari, K. Rikimaru, Y. Morohashi, T. Kan, H. Natsugari, T. Fukuyama, T. Tomita, T. Iwatsubo, J. Bio. Chem., 278, 18664 (2003).
34) H. F. Dovey, V. John, J. P. Anderson, L. Z. Chen, P. de Saint Andrieu, L. Y. Fang, S. B. Freedman, B. Folmer, E. Goldbach, E. J. Holsztynska, K. L. Hu, K. L. Johnson-Wood, S. L. Kennedy, D. Kholodenko, J. E. Knops, L. H. Latimer, M. Lee, Z. Liao, I. M. Lieberburg, R. N. Motter, L. C. Mutter, J. Nietz, K. P. Quinn, K. L. Sacchi, P. A. Seubert, G. M. Shopp, E. D. Thorsett, J. S. Tung, J. Wu, S. Yang, C. T. Yin, D. B. Schenk, P. C. May, L. D. Altstiel, M. H. Bender, L. N. Boggs, T. C. Britton, J. S. Clemens, D. L. Czilli, D. K. Dieckman-MacGinty, J. J. Droste, K. S. Fuson, B. D. Gitter, P. A. Hyslop, E. M. Johnstone, W. Y. Li, S. P. Little, T. E. Mabry, F. D. Miller, J. E. Audia, J. Neurochem, 76, 173 (2001).
35) T. Kan, Y. Tominari, Y. Morohashi, H. Natsugari, T. Tomita, T. Iwatsubo, T. Fukuyama, Chem. Commun.
2003, 2244.
36) N. Takasugi, T. Tomita, I. Hayashi, M. Niimura, Y. Takahashi, G. Thinakaram, T. Iwastubo, Nature, 422, 438 (2003).
37) フォトアフィニティラベリングの総説 : a) F. Kotozyba-Hilbert, I. Kapfer and M. Goeldner, Angew.
Chem., Int. Ed. Engl., 34, 1296 (1995). b) G. Dorman and G. D. Prestwich, Biochemistry, 33, 5661 (1994).
38) Y. Morohashi, T. Kan, Y. Tominari, H. Fuwa, Y. Okamura, N. Watanabe, H. Natsugari, T. Fukuyama, T. Iwatsubo, T. Tomita, submitted.
(Manuscript received Aug. 2004) (Revision received Aug. 2005)
執筆者紹介
菅 敏幸
(Toshiyuki Kan)
静岡県立大学 薬学部 薬品製造化学教室 教授 [ご経歴] 1986年 北海道大学理学部化学科卒業,1993年 同大学大学院理学研究科博士課程 修了(指導教官,白濱晴久),財団法人サントリー生物有機科学研究所研究員(1993-1996), 1996年 東京大学薬学部助手,2004年 東京大学大学院薬学系研究科助教授,2005年より現職。 2002年 有機合成化学協会奨励賞,2004年 日本薬学会薬学研究ビジョン部会賞受賞。 [ご専門] 有機合成化学,化学生物学福山 透
(Tohru Fukuyama)
東京大学 大学院薬学系研究科 教授 [ご経歴] 1971年 名古屋大学農学部農芸化学科卒業,1977年 ハーバード大学化学科Ph.D.取 得,1977-1978年 同大学大学院博士研究員,1978年 ライス大学化学科助教授,1982年 同准 教授,1988年 同教授,1995年 東京大学薬学部教授,1997年より現職。1993年 アメリカ化学会Arthur C. Cope Scholar Award,2001年 有機合成化学協会賞,2003
年 国際複素環学会Senior Award,2004年 アメリカ化学会Arthur C. Cope Award受賞。
寄稿論文 関連製品
ニトロベンゼンスルホニル化合物 ほか
X SO2Cl Y X SO2NH2 Y NO2 SO2NHR H2N n NH2 n SO2NH NO2 NH2 p.3-4X=NO2, Y=H : NsCl (11a)
500g 21,800円25g 2,250円[N0142] X=H, Y=NO2 : pNsCl (11b) 25g 9,600円 5g 3,500円[N0144] p.4-5 X=NO2, Y=H : NsNH2 (18) 25g 21,100円 5g 7,150円[N0705] X=H, Y=NO2 5g 7,350円[N0697] p.4-5 R=Boc (19a) 5g 28,000円 1g 8,500円[B2303] R=Alloc (19b) 5g 29,500円[A1632] R=Cbz (19c) 5g 25,500円[C1757] p.6 Diamine derivatives n=1 500ml 4,900円25ml 1,400円[D0114] n=2 500g 9,300円 25g 1,600円[D0239] n=3 25ml 16,300円 5ml 6,050円[D0108] n=4 500g 4,300円 25g 1,400円[D0095] n=5 25g 12,900円 5g 4,800円[D0094] n=6 25g 4,800円[D0107] n=7 25g 13,600円 5g 4,800円[D0106] n=8 25g 8,250円[D0085] n=9 5g 13,000円 1g 4,500円[D2075] n=10 500g 35,300円 25g 4,300円[D0091] n=0 1g 12,400円[A1627] n=1 (27) 1g 12,400円[A1628] n=2 (28) 1g 12,400円[A1630] n=3 •HCl 1g 21,500円[A1661] n=4 •HCl 1g 21,500円[A1662]